自2012年1月20日,《国家药品安全“十二五”规划》首次提出仿制药质量一致性评价至今已有四年时间,该工作一度搁浅,于2015年底再度启动。为帮助企业更好的开展工作,本文梳理了相关政策的出台历程及要点。
2012年1月20日,国务院印发了《国家药品安全“十二五”规划》 。
“规划”要求对2007年修订的《药品注册管理办法》施行前批准的仿制药,分期分批与被仿制药进行质量一致性评价,其中纳入国家基本药物目录、临床常用的仿制药在2015年前完成,未通过质量一致性评价的不予再注册,注销其药品批准证明文件。药品生产企业必须按《药品注册管理办法》要求,将其生产的仿制药与被仿制药进行全面对比研究,作为申报再注册的依据。
CFDA依次于2012年11月22日发布了《仿制药质量一致性评价工作方案(征求意见稿)》与2013年7月11日发布了《国家食品药品监督管理局关于开展仿制药质量一致性评价工作的通知》(正式稿),两者的工作计划对比如下:
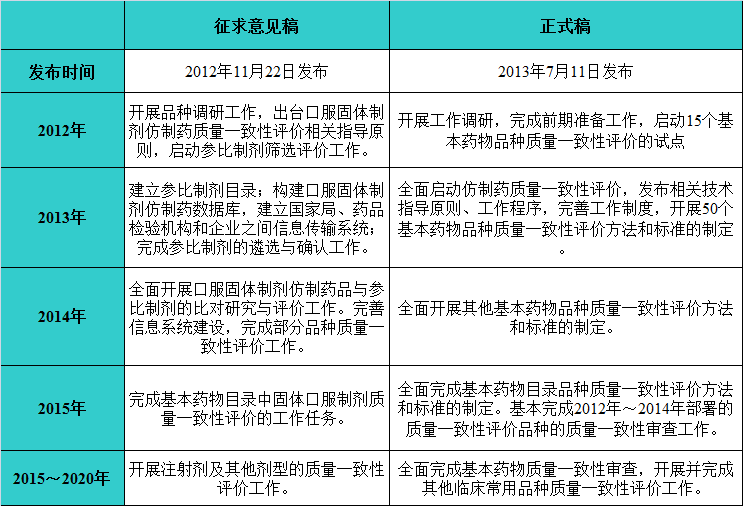
经过对比,尤其从两稿的第四点(即2015年工作计划)可以看出,正式稿在完成目标上较征求意见稿做了较大妥协。
2013年07月11日, CFDA发布《国家食品药品监督管理总局办公厅关于2013年度仿制药质量一致性评价方法研究任务的通知》。
通知对各省、自治区、直辖市食品药品监督管理局(药品监督管理局)、中国食品药品检定研究院就2013年度仿制药质量一致性评价方法研究任务作出了安排。
2014年01月29日,CFDA发布《国家食品药品监督管理总局公开征求仿制药质量一致性评价有关指导原则等意见》。
中国食品药品检定研究院公示了3个拟评价品种的评价方法和《普通口服固体制剂溶出曲线测定与比较指导原则(征求意见稿)》、《口服固体制剂参比制剂确立原则(征求意见稿)》2个技术指导原则。
2015年08月18日,国务院发布《国务院关于改革药品医疗器械审评审批制度的意见》。
其中主要目标的前三条均直接或间接和仿制药质量一致性评价相关: 
意见中明确提出要通过仿制药质量一致性评价提高仿制药质量。提高药品审批标准的方法之一是“将仿制药由现行的‘仿已有国家标准的药品’调整为‘仿与原研药品质量和疗效一致的药品’”,即广义的仿制药申报均应严格按照仿制药质量一致性评价的要求进行研究。另外,这个要求也意味着提高了研发成本和周期,有助于解决注册申请积压问题。
2015年10月30日,CFDA发布《食品药品监管总局办公厅关于征求普通口服固体制剂参比制剂选择和确定指导原则等意见的通知》。再次公布《普通口服固体制剂参比制剂选择和确定指导原则(征求意见稿)》《普通口服固体制剂溶出曲线测定与比较指导原则(征求意见稿)》《仿制药质量一致性评价人体生物等效性研究技术指导原则(征求意见稿)》。
2015年11月06日,CFDA发布《关于征求药品上市许可持有人制度试点方案和化学药品注册分类改革工作方案两个征求意见稿意见的公告》。
其中很重要的调整是“新药是指未在中国境内外上市销售的药品,将境外上市境内未上市药品纳入仿制药。”。调整之前“境外上市境内未上市药品”按新药管理,虽然这类新药之前也要求与原研进行质量对比研究,但是研究的内容和实质要求与现在所说的仿制药质量一致性评价还是有很大差别。
2015年11月18日,为贯彻落实《国务院关于改革药品医疗器审评审批制度的意见》提出的开展仿制药质量和疗效一致性评价工作的相关要求,CFDA发布《国家食品药品监督管理总局关于征求〈关于开展仿制药质量和疗效一致性评价的意见(征求意见稿)〉意见的公告》。
征求意见稿中以下内容比较引人关注:
1、明确评价对象和时限
对已经批准上市的仿制药,凡没有按照与原研药质量和疗效一致的原则审批的,均需按照上述原则开展一致性评价。对2007年10月1日前批准的国家基本药物目录(2012年版)中化学药品仿制药口服固体制剂,应在2018年底之前完成一致性评价,届时没有通过评价的,注销药品批准文号。
对2007年以前批准上市的其他仿制药品和2007年以后批准上市的仿制药品,自首家品种通过一致性评价后,其他生产企业的相同品种在3年内仍未通过评价的,注销药品批准文号。
这意味着所有已上市的仿制药均纳入了一致性评价的范围,涉及品种大大超过了2012/2013年CFDA制定的工作计划。
2、合理选用研究方法
原则上企业应采用体内生物等效性试验的方法进行评价,允许企业采取体外溶出度试验的方法进行评价。采用体外溶出度试验方法进行评价的品种,以后还应当采取体内生物等效性试验的方法进行后续评价。开展体内生物等效性试验时,企业应根据仿制药生物等效性试验的有关规定组织实施。对无参比制剂的,企业需按照有关要求进行临床有效性试验。
这意味着体内生物等效性试验成为评价是否一致的金标准,与2012/2013年版本发生了重大改变。
3、鼓励企业开展一致性评价工作
通过一致性评价的品种,由食品药品监管总局向社会公布。企业可在药品说明书、标签中予以标示体内评价和体外评价的标识;企业可以申报作为该品种的药品上市许可持有人,委托其他药品生产企业生产,并承担上市后的相关法律责任。通过一致性评价的品种,社保部门在医保支付方面予以适当支持。医疗机构优先采购并在临床中优先选用。发展改革委、工业和信息化部对通过一致性评价药品企业的技术改造给予支持。
同一品种达到3家以上通过一致性评价的,在集中采购等方面不再选用未通过评价的品种。
这条意见不仅表明通过一致性评价品种可享受一系列扶持政策,同样也再次说明本次一致性评价工作涉及品种范围之广以及不通过的后果之严重。
由上文可以看出,仿制药质量一致性评价不仅仅是一项针对已上市产品的质量再评价工作,还将直接影响“化学药品注册分类改革工作”和“解决注册申请积压工作”。后两项工作能否取得预期效果,与仿制药质量一致性评价进展情况密切相关。因此仿制药质量一致性评价是势在必行的一项工作,是中国制药工业整体水平和医疗服务水平提高的必然要求。
当然,征求意见稿中还有一些内容争议较大,最终也有可能在正式意见中进行调整,如第一阶段基药完成的时间期限,以及对2007年前后批准的产品的政策等。
此外,即便体内生物等效性试验成为评价是否一致的金标准,用体外溶出曲线的研究来指导处方工艺的调整,仍是开展一致性评价工作的前提条件和必经之路,且征求意见稿中也明确提出“允许企业采取体外溶出度试验的方法进行评价。采用体外溶出度试验方法进行评价的品种,以后还应当采取体内生物等效性试验的方法进行后续评价。”即使是由于各种原因开展BE试验有困难,也不应影响企业开展一致性评价的工作。
不管怎样,一致性评价工作的开展已经是箭在弦上,一触即发。相关企业应做好准备,提前开展溶出曲线、处方工艺等药学研究,做到有备无患,才能高枕无忧。
(本文由国健医药咨询注册中心供稿,转载请注明来源。)




{kind=link}